т. 20, № 2, стр. 100-113 (1994)
Реакция окси- и аминоацильного включения
А.М. Шкроб
Институт биомедицинской химии РАМН, Москва
|
т. 20, № 2, стр. 100-113 (1994) |
| © А.М. Шкроб |
Реакция окси- и аминоацильного включенияА.М. Шкроб Институт биомедицинской химии РАМН, Москва |
Человечество возникло и имеет шанс уцелеть благодаря своей способности передавать от поколения к поколению накопленный рациональный и нравственный опыт. И если в ветхозаветное время этот опыт люди приобретали главным образом в семье, то уже не одно тысячелетие вместе с родителями или после них нас воспитывают учителя. Чти родителей и учителей своих! - таков глубинный смысл едва ли не главной из заповедей, переданных Моисеем своему народу.
* * *
* * *
* * *
* * *
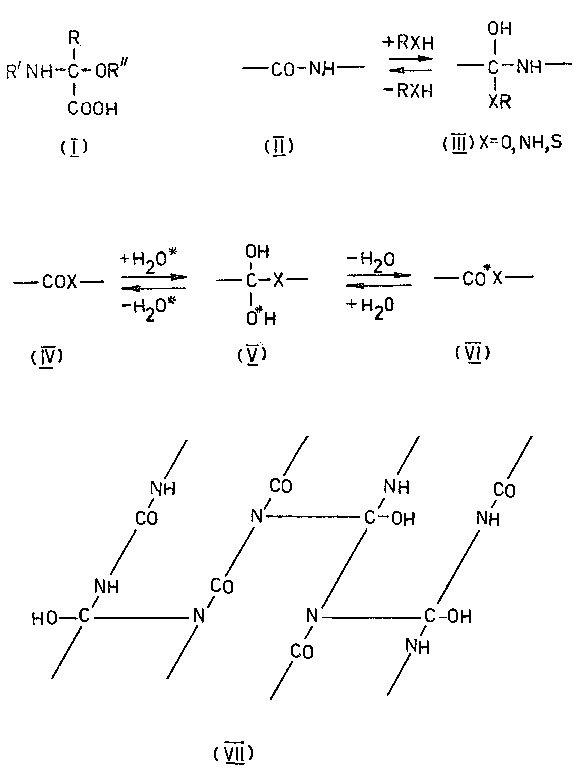
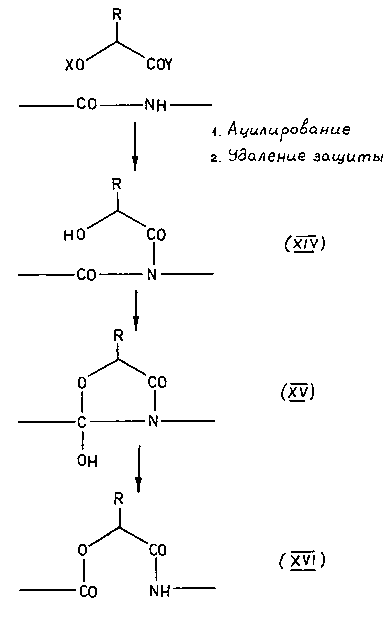
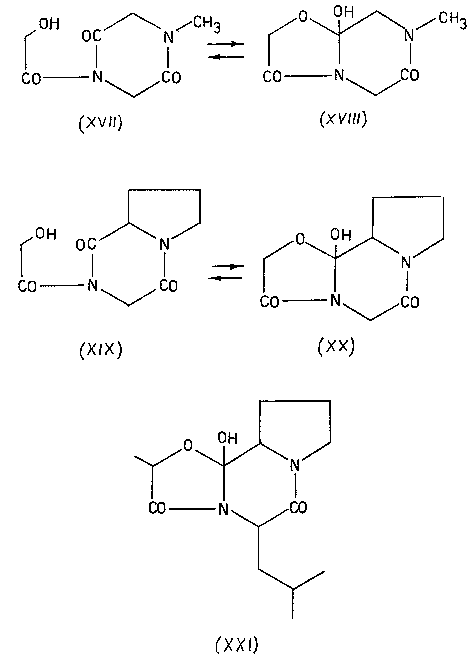
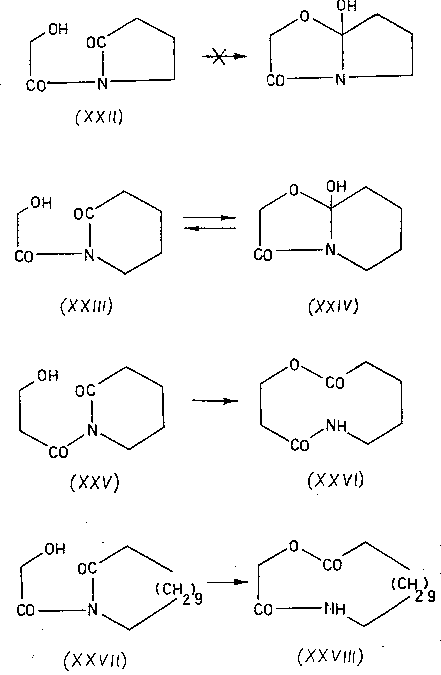
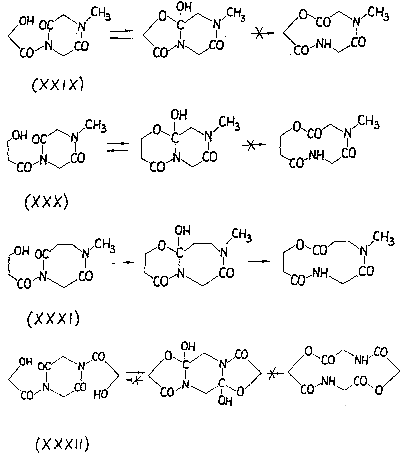
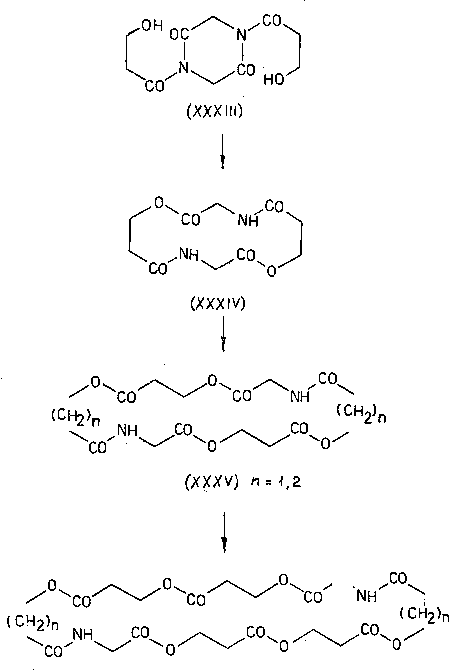
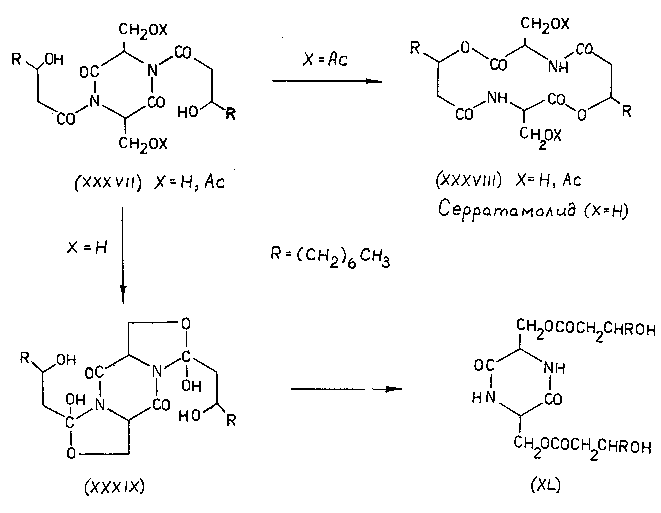
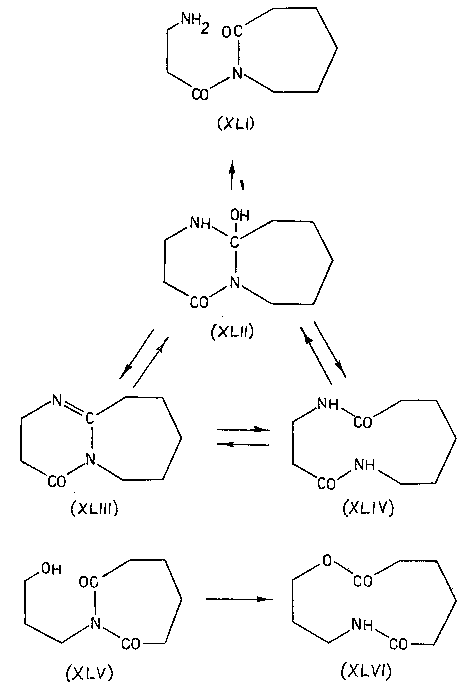
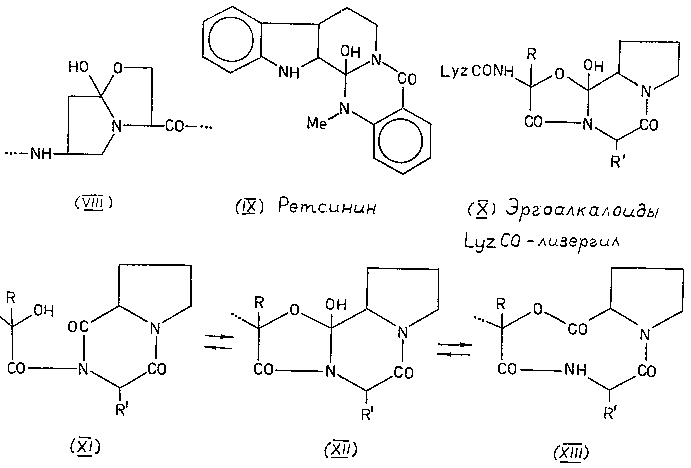
После этого осталось только окончательно упростить ситуацию и, тем самым, превратить экзотическую изомеризацию в универсальный синтетический прием. Этот шаг - исключение цикла и переход к линейным моделям, разумеется, также был вскоре сделан [45-48], хотя, повторяю, сейчас я просто не понимаю, почему с него не начали.
* * *
* * *
* * *
* * *
* * *
Список литературы
1. Шемякин М.М., Чаман Е.С. // ЖОХ. 1955. Т. 25. N 7. С. 1360-1365.
2. Шемякин М.М., Чаман Е.С., Лурье С.И. // Журн. общ. химии. 1955. Т. 25. N 9. С. 1799-1802.
3. Шемякин М.М., Чаман Е.С., Денисова Л.И. // Докл. АН СССР. 1956. Т. 106.
N4. С. 675-678.
4. Шемякин М.М., Денисова Л.И., Чаман Е.С. // Изв. АН СССР, Сер. хим. 1959. N 4. С. 690-694.
5. Шемякин М.М., Антонов В.К. // Докл. АН СССР. 1959. Т. 129. N 2. С. 349-350.
6. Shemyakin M.M., Tschaman E.S., Ravdel G.A., Rodionov W.I. // Bull. Soc. chim. France. 1959. N 3. P. 530-539.
7. Shemyakin M.M. // Angew. Chem. 1960. V. 72. N 10. P. 342-345; Успехи химии. 1962. Т. 31. N 3. С. 269-284.
8. Bender M.L. // J. Amer. Chem. Soc. 1953. V. 75. P. 5986.
9. Bender M.L. // Chem. Rev. 1960. V. 60. N 1. P. 54.
10. Frank F.C. // Nature. 1936. V. 138. P. 242.
11. Wrinch D. // Nature 1957. V. 179. P. 536; 1962. V. 193. P. 245; 1963. V. 199. P. 564 (см. также цит. там работы).
12. Wrinch D. // Chemical Aspects of the Structure of Small Peptides. 1960. E. Munksgaard Publ. Co. Copenhagen.
13. Pauling L.C., Niemann C. // J. Amer. Chem. Soc. 1939. V. 61. P. 1860.
14. Edward J.T. // Research. 1955. V. 8. P. 38.
15. Bernhard S.A. // J. Cell. Comp. Physiol. 1959. V. 54 (Suppl. 1). P. 135.
16. Asahina Y., Ohta T // J. Pharm. Soc. Japan. 1926. V. 53. P. 293.
17. Ohta T. // J. Pharm. Soc. Japan. 1945. V. 65B. P. 89.
18. Chatterjee A., Bose S., Ghosh C. // Tetrahedron. 1959. V. 7. P. 257.
19. Pachter I.J., Suld G. // J. Org. Chem. 1960. V. 25. P. 1680.
20. Pachter I.J., Mohrbacher R.J., Zacharias P. // J. Amer. Chem. Soc. 1961. V. 83. P. 635.
21. Stoll A., Hofmann A., Petrzilka Th. // Helv. chim. acta. 1951. V. 34. P. 1544.
22. Stoll A., Hofmann A. // Helv. chim. acta. 1950. V. 33. P. 1705.
23. Stoll A., Petrzilka Th., Becker B. // Helv. chim. acta. 1950. V. 33. P. 57.
24. Grob C.A., Meier W. // Helv. chim. acta. 1956. V. 39. P. 776.
25. Stoll A., Hofmann A., Leemann H.G., Ott H., Schlenk H.R. // Helv. chim. acta. 1956. V. 39. P. 1165.
26. Green M., Lucken E. // Helv. chim. acta. 1961. V. 44. P. 1417.
27. Antonov V.K., Ravdel G.A., Shemyakin M.M. // Chimia. 1960. V. 14. P. 374.
28. Равдель Г.А., Крит Н.А., Щукина Л.А., Шемякин М.М. // Докл. АН СССР. 1961. Т. 137. С. 1377.
29. Hofmann A., Frey A.J., Ott H. // Experientia. 1961. V. 17. P. 206.
30. Sheppard R.C. // Experientia. 1963. V. 19. P. 125.
31. Shemyakin M.M., Aldanova N.A., Vinogradova E.I., Feigina M.Yu. // Tetrahedron Lett. 1963. N 28. P. 1921-1925.
32. Hoffmann A., Frey A.J., Ott H., Rutschmann H. // Тез. сообщ. на 4-м Европейском симпозиуме по пептидам (Москва, авг. 1961); Журн. Всесоюзн. хим. общ. им. Д.И. Менделеева. 1962. Т. 7. С. 468.
33. Hofmann A., Ott H., Griot R., Stadler P.A., Frey A.J. // Helv. chim. acta 1963. V. 46. P. 2306.
34. Ott H., Frey A.J., Hofmann A.H. // Tetrahedron. 1963. V. 19. P. 1675.
35. Antonov V.K., Shkrob A.M., Shemyakin M.M. // Peptides (Procs of the 6th European Symp. on Peptides, Oxford, Sept. 1962) / Ed. Young G.T. Pergamon Press. 1963. P. 221-228.
36. Shemyakin M.M., Antonov V.K., Shkrob A.M., Sheinker Yu.N., Senyavina L.B. // Tetrahedron Lett. 1962. N 16. P. 701-707.
37. Антонов В.К., Шкроб А.М., Шемякин М.М. // Журн. общ. химии. 1967. Т. 37. N 10. С. 2225-2233.
38. Antonov V.K., Shkrob A.M., Shemyakin M.M. // Tetrahedron Letters. 1963. N 7. P. 439-443.
39. Антонов В.К., Шкроб А.М., Шемякин М.М. // Журн. общ. химии. 1965. Т. 35. N 8. С. 1380-1389.
40. Wieland Th., Urbach H. // Liebig's Ann. Chem. 1958. V. 613. P. 84.
41. Гольдфарб Я.Л., Беленький Л.И. // Успехи химии. 1960. Т. 29. С. 470.
42. Sebenda J., Stehlicek J. // Collect. Czech. Chem. Communs. 1963. V. 28. P. 2731.
43. Sebenda J., Mikulova B. // Collect. Czech. Chem. Communs. 1964. V. 29. P. 738.
44. Антонов В.К., Шкроб А.М., Крылова Ю.И., Шемякин М.М. // Журн. общ. химии. 1966. Т. 36. N 6. С. 1008-1012.
45. Shemyakin M.M., Antonov V.K. // Pure Appl. Chem. 1964. V. 9. N 1. P. 75-94.
46. Shemyakin М.M., Antonov V.K., Shkrob A.M., Shchelokov V.I., Agadzhanyan Z.E. // Tetrahedron. 1965. V. 21. P. 3537-3572.
47. Shemyakin M.M., Antonov V.K. // Acta chim. Hung. 1965. V. 44. N 1-2. P. 93-98.
48.Brenner M., Zimmerman J.P., Wehrmuller J., Quitt P., Hartmann A., Schneider W., Beglinger U. // Helv. chim. acta. 1957. V. 40. P. 1497.
49. Stich K., Leemann H.G. // Helv. chim. acta. 1963. V. 46. P. 1151.
50. Frey A.J., Griot R.G. // Tetrahedron. 1963. V. 19. P. 1161.
51. Шкроб А.М., Крылова Ю.И., Антонов В.К., Шемякин М.М. // Журн. общ. химии. 1965. Т. 35. N 8, С. 1389-1398.
52. Антонов В.К., Андреева Л.И., Лякишева А.Г., Шемякин М.М. // Журн. общ. химии. 1967. Т. 37. N 8, С. 1703-1707.
53. Шкроб А.М., Крылова Ю.И., Антонов В.К., Шемякин М.М. // Изв. АН СССР, Сер. хим. 1964. N 4. С. 774.
54. Hall H.K., Brandt M.K., Mason R.M. // J. Amer. Chem. Soc. 1958. V. 80. P. 6420.
55. Lee C.M., Kumler W.D. // J. Amer. Chem. Soc. 1962. V. 84. P. 565.
56. Antonov V.K., Shkrob A.M., Shchelokov V.I., Shemyakin M.M. // Tetrahedron Lett. 1963. N 21. P. 1353-1360.
57. Антонов В.К., Щелоков В.И., Шемякин М.М. // Изв. АН СССР (сер. хим.). 1963. N 6. С. 1145; Журн. общ. химии. 1965. Т. 35. N 12. 2239-2246.
58. Shemyakin M.M., Ovchinnikov Yu.A., Antonov V.K., Kiryushkin A.A., Ivanov V.T., Shchelokov V.I., Shkrob A.M. // Tetrahedron Lett. 1964. N 1. P. 47-54; Изв. АН СССР, Сер. хим. 1963. N 12. С. 2233.
59. Antonov V.K., Agadzhanyan Ts.E., Telesnina T.R., Dvoryantzeva G.G., Sheinker Yu.N., Shemyakin M.M. // Tetrahedron Lett. 1964. N 13. P. 727-732.
60. Антонов В.К., Агаджанян Ц.Е., Телеснина Т.Р., Шемякин М.М. // Журн. общ. химии. 1965. Т. 35. N 12. С. 2231-2238.
61. Rothe M., Steinberger R. // Angew. Chem. 1968. V. 21. P. 909-910.
62. Glover G.I., Rappoport H. // J. Amer. Chem. Soc. 1964. V. 86. P. 3398.
63. Антонов В.К., Моргулян В.С., Шемякин М.М. // Журн. общ. химии. 1967. Т. 37. N 7. С. 1597-1600.
64. Shkrob A.M., Krylova Yu.I., Antonov V.K., Shemyakin M.M. // Tetrahedron Lett. 1967. N 28. P. 2701-2706; Журн. общ. химии. 1968. Т. 38. N 9. С. 2030-2046, 2046-2050, 2051-2064.

А.М. Шкроб,119571 Москва, ул. 26-ти Бакинских комиссаров
3-3-448
телефон 7-(095)-433-37-09
![]()